Biopython
Biopython նախագիծը հաշվարկային կենսաբանության և կենսաինֆորմատիկայի ոչ առևտրային նպատակով Python գործիքների բաց կոդով շարք է, որը ստեղծվել է մշակողների միջազգային ասոցիացիայի կողմից[1][2][3]։ Այն պարունակում է կլասեր, որոնք ներկայացնում են կենսաբանական հաջորդականությունները և հաջորդականությունների ծանոթագրությունները, և ունակ է կարդալ և գրել տարբեր ֆայլի ձևաչափեր։ Այն նաև հնարավորություն է տալիս ծրագրային միջոցներով մուտք գործել կենսաբանական տեղեկատվության առցանց տվյալների շտեմարաններ, ինչպես օրինակ ՝ NCBI: Առանձին մոդուլները տարածում են Biopython- ի հնարավորությունները հաջորդականության դասավորության, սպիտակուցների կառուցվածքի, պոպուլյացիոն գենետիկայի, ֆիլոգենետիկայի, հաջորդականության մոտիվների և մեքենայական ուսուցման վրա։ Biopython- ը մի շարք Bio* նախագծերից մեկն է, որը նախատեսված է հաշվարկային կենսաբանության մեջ ծածկագրերի կրկնօրինակումը նվազեցնելու համար[4]։
Պատմություն
Biopython- ի զարգացումը սկսվել է 1999 թ. և այն առաջին անգամ թողարկվել է 2000 թ. հուլիսին[5]։ Այն մշակվել է նույն ժամանակահատվածում և այլ նախագծերի միևնույն նպատակներով, որոնք կենսաինֆորմատիկային հնարավորություն են տվել ունենալ համապատասխան ծրագրավորման լեզուներ, ներառյալ BioPerl- ը, BioRuby- ն և BioJava- ն։ Նախագծի մշակողները ներառում էին Ջեֆ Չանգը, Էնդրյու Դալկեն և Բրեդ Չեփմանը, չնայած մինչ օրս ավելի քան 100 մարդ ներդրում է կատարել այս նախագծում[6]։ 2007 թվականին ստեղծվեց նման Python նախագիծ, այն է ՝ PyCogent[7]:
Biopython- ի սկզբնական տարբերակը ներառում էր կենսաբանական հաջորդականության ֆայլերի մուտք, ինդեքսավորում և մշակում։ Թեև սա դեռևս հիմնական ուշադրության կենտրոնում է, հաջորդ տարիների ընթացքում մոդուլների ավելացումն ընդլայնել է իր ֆունկցիոնալությունը `ընդգրկելով կենսաբանության լրացուցիչ ոլորտներ։
1.77 տարբերակի դրությամբ Biopython- ն այլևս չի աջակցում Python 2 -ին[8]։
Դիզայն
Հնարավորության դեպքում Biopython- ը հետևում է Python ծրագրավորման լեզվի կողմից օգտագործվող պայմանականություններին `Python- ին ծանոթ օգտվողների համար ավելի դյուրին դարձնելու համար։ Օրինակ ՝ Seq և SeqRecord օբյեկտները կարող են շահարկվել slicing-ի միջոցով, ինչպես Python- ի strings և lists: Այն նաև նախագծված է գործառականորեն նման Bio* այլ նախագծերին, օրինակ ՝ BioPerl- ին[5]։
Biopython- ն ունակ է կարդալ և գրել ամենատարածված ֆայլի ձևաչափերն իր ֆունկցիոնալ ոլորտներից յուրաքանչյուրի համար, և դրա լիցենզիան թույլատրելի է և համատեղելի է ծրագրային ապահովման այլ լիցենզիաների հետ, ինչը թույլ է տալիս Biopython-ն օգտագործել տարբեր ծրագրային նախագծերում[3]։
Հիմնական առանձնահատկություններ և օրինակներ
Հաջորդականություն
Biopython- ի հիմնական հասկացությունը կենսաբանական հաջորդականությունն է, և դա ներկայացված է Seq կլասով[9]։ Biopython Seq օբյեկտը շատ առումներով նման է Python- ի string-ին՝ այն աջակցում է Python slice նշումը, կարող է զուգակցվել այլ հաջորդականությունների հետ և անփոփոխ է։ Բացի այդ, այն ներառում է հաջորդականության համար հատուկ մեթոդներ և հստակեցնում է օգտագործվող հատուկ կենսաբանական այբուբենը։
>>> # This script creates a DNA sequence and performs some typical manipulations
>>> from Bio.Seq import Seq
>>> dna_sequence = Seq('AGGCTTCTCGTA', IUPAC.unambiguous_dna)
>>> dna_sequence
Seq('AGGCTTCTCGTA', IUPACUnambiguousDNA())
>>> dna_sequence[2:7]
Seq('GCTTC', IUPACUnambiguousDNA())
>>> dna_sequence.reverse_complement()
Seq('TACGAGAAGCCT', IUPACUnambiguousDNA())
>>> rna_sequence = dna_sequence.transcribe()
>>> rna_sequence
Seq('AGGCUUCUCGUA', IUPACUnambiguousRNA())
>>> rna_sequence.translate()
Seq('RLLV', IUPACProtein())
Հաջորդականության ծանոթագրություն
SeqRecord կլասը նկարագրում է հաջորդականությունները ՝ տեղեկատվության հետ միասին, ինչպիսիք են անունը, նկարագրությունը և հատկանիշները ՝ SeqFeature օբյեկտների տեսքով։ Յուրաքանչյուր SeqFeature օբյեկտ սահմանում է հատկության տեսակը և դրա գտնվելու վայրը։ Առանձնահատկությունների տեսակները կարող են լինել «գեն», «CDS» (կոդավորման հաջորդականություն), «կրկնվող_հատված», «բջջային_էլեմենտ» կամ այլ, իսկ հաջորդականության մեջ հատկանիշների դիրքը կարող է լինել ճշգրիտ կամ մոտավոր։
>>> # This script loads an annotated sequence from file and views some of its contents.
>>> from Bio import SeqIO
>>> seq_record = SeqIO.read('pTC2.gb', 'genbank')
>>> seq_record.name
'NC_019375'
>>> seq_record.description
'Providencia stuartii plasmid pTC2, complete sequence.'
>>> seq_record.features[14]
SeqFeature(FeatureLocation(ExactPosition(4516), ExactPosition(5336), strand=1), type='mobile_element')
>>> seq_record.seq
Seq('GGATTGAATATAACCGACGTGACTGTTACATTTAGGTGGCTAAACCCGTCAAGC...GCC', IUPACAmbiguousDNA())
Մուտք և ելք
Biopython- ը կարող է կարդալ և գրել մի շարք սովորական հաջորդական ձևաչափերով, այդ թվում `FASTA, FASTQ, GenBank, Clustal, PHYLIP և NEXUS: Ֆայլեր կարդալիս ֆայլում նկարագրական տեղեկատվությունը օգտագործվում է Biopython կլասերի անդամներին համալրելու համար, օրինակ ՝ SeqRecord: Սա թույլ է տալիս մեկ ֆայլի ձևաչափի գրառումները փոխակերպել մյուսների։
Շատ մեծ հաջորդականությամբ ֆայլերը կարող են գերազանցել համակարգչի հիշողության ռեսուրսները, ուստի Biopython- ը մեծ գրառումներով ֆայլեր մուտք գործելու տարբեր տարբերակներ է տրամադրում։ Նրանք կարող են ամբողջությամբ բեռնվել հիշողության մեջ Python տվյալների կառուցվածքներում, օրինակ ՝ ցուցակներում կամ բառարաններում, ապահովելով արագ մուտք հիշողության օգտագործումով։ Այլապես, ֆայլերը կարող են կարդացվել սկավառակից, ըստ անհրաժեշտության, ավելի դանդաղ կատարմամբ, բայց հիշողության ցածր պահանջներով։
>>> # This script loads a file containing multiple sequences and saves each one in a different format.
>>> from Bio import SeqIO
>>> genomes = SeqIO.parse('salmonella.gb', 'genbank')
>>> for genome in genomes:
... SeqIO.write(genome, genome.id + '.fasta', 'fasta')
Առցանց տվյալների շտեմարանների մուտք
Bio.Entrez մոդուլի միջոցով Biopython- ի օգտվողները կարող են ներբեռնել կենսաբանական տվյալները NCBI տվյալների բազաներից։ Entrez որոնման համակարգի տրամադրած գործառույթներից յուրաքանչյուրը հասանելի է այս մոդուլի գործառույթների միջոցով, ներառյալ գրառումների որոնումը և ներբեռնումը։
>>> # This script downloads genomes from the NCBI Nucleotide database and saves them in a FASTA file.
>>> from Bio import Entrez
>>> from Bio import SeqIO
>>> output_file = open('all_records.fasta', "w")
>>> Entrez.email = 'my_email@example.com'
>>> records_to_download = ['FO834906.1', 'FO203501.1']
>>> for record_id in records_to_download:
... handle = Entrez.efetch(db='nucleotide', id=record_id, rettype='gb')
... seqRecord = SeqIO.read(handle, format='gb')
... handle.close()
... output_file.write(seqRecord.format('fasta'))
Ֆիլոգենիա

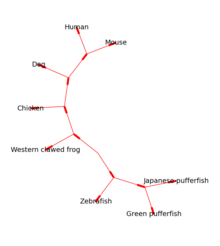
Bio.Phylo մոդուլը տրամադրում է գործիքներ ֆիլոգենետիկ ծառերի հետ աշխատելու և դրանք տեսանելի դարձնելու համար։ Կարդալու և գրելու համար ապահովվում են տարբեր ֆայլի ձևաչափեր, այդ թվում ՝ Newick, NEXUS և phyloXML: Tree և Clade օբյեկտների միջոցով կատարվում են ծառերի սովորական մանիպուլյացիաները և անցումները։ Օրինակները ներառում են ծառերի ֆայլերի փոխակերպում և համադրում, ծառից ենթախմբերի դուրսբերում, ծառի արմատը փոխելը և ճյուղի առանձնահատկությունների վերլուծություն, ինչպիսիք են երկարությունը կամ միավորը[11]։
Արմատացած ծառերը կարող են գծվել ASCII- ով կամ matplotlib- ի միջոցով (տես նկար 1), իսկ Graphviz գրադարանը կարող է օգտագործվել առանց արմատավորված դասավորություններ ստեղծելու համար (տես նկար 2)։
Գենոմային դիագրամներ
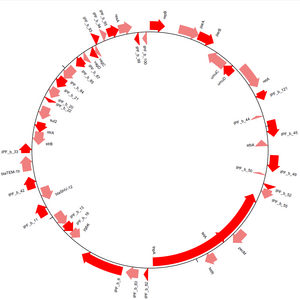
GenomeDiagram մոդուլն ապահովում է Biopython- ի շրջանակներում հաջորդականությունների տեսանելի մեթոդներ[13]։ Հերթականությունները կարող են գծվել գծային կամ շրջանաձև տեսքով (տես նկար 3), ինչպես նաև բազմաթիվ ելքային ձևաչափեր են ապահովված, ներառյալ PDF և PNG: Դիագրամները ստեղծվում են ՝ հետքեր պատրաստելով, այնուհետև հաջորդական հատկանիշներ ավելացնելով այդ հետքերին։ Մեկը, շրջանակելով հաջորդականության առանձնահատկությունները և օգտագործելով դրանց հատկանիշները որոշելու համար, թե արդյոք դրանք ինչպես են ավելացվում գծապատկերի հետքերում, կարող է մեծ վերահսկողություն հաստատել վերջնական դիագրամի արտաքին տեսքի վրա։ Խաչաձև կապերը կարող են կազմվել տարբեր հետքերի միջև ՝ թույլ տալով մեկին համեմատել մի քանի հաջորդականություններ մեկ դիագրամում։
Մակրոմոլեկուլյար կառուցվածք
Bio.PDB մոդուլը կարող է բեռնել մոլեկուլային կառուցվածքներ PDB և mmCIF ֆայլերից և ավելացվել է Biopython- ին 2003 թվականին։ Կառուցվածքի օբյեկտը կենտրոնականն է այս մոդուլի համար և հիերարխիկ կերպով կազմակերպում է մակրոմոլեկուլային կառուցվածքը. Structure, որոնք պարունակում են Modelօբյեկտներ, որոնք էլ պարունակում են Chain օբյեկտներ, սրանք էլ պաուրնակում են Residue օբյեկրներ և վերջինն էլ իր հերթին պաուրնակում է Atom օբյեկտներ։ Ոչ ճիշտ հաջորդականությամբ մնացորդներն ու ատոմները ստանում են իրենց կլասերը ՝ DisorderedResidue և DisorderedAtom, որոնք նկարագրում են իրենց անորոշ դիրքերը։
Օգտագործելով Bio.PDB- ն ՝ մեկը կարող է ղեկավարել մակրոմոլեկուլային կառուցվածքի ֆայլի առանձին բաղադրիչներ, օրինակ ՝ սպիտակուցի յուրաքանչյուր ատոմի հետազոտումը։ Հնարավոր են ընդհանուր վերլուծություններ, օրինակ ՝ հեռավորությունների կամ անկյունների չափում, մնացորդների համեմատություն և մնացորդների խորության հաշվարկ։
Պոպուլյացիոն գենետիկա
Bio.PopGen մոդուլն ավելացնում է Biopython- ի հնարավորւոթյունը Genepop- ի համար, որը ծրագրային փաթեթ է `պոպուլյացիոն գենետիկայի վիճակագրական վերլուծության համար[14]։ Սա թույլ է տալիս վերլուծել Հարդի -Վայնբերգի հավասարակշռությունը, կապի անհավասարակշռությունը և պոպուլյացիոն ալելների ալելի շատ հաճախականությունների այլ առանձնահատկությունները։
Այս մոդուլը կարող է նաև իրականացնել պոպուլյացիայի գենետիկական սիմուլյացիա ՝ օգտագործելով fastsimcoal2 ծրագրի հետ համատեղվող տեսությունը[15]։
Տես նաև
- BioPerl
- BioRuby
- BioJS
- BioJava
Ծանոթագրություններ
- ↑ Chapman, Brad; Chang, Jeff (August 2000). «Biopython: Python tools for computational biology». ACM SIGBIO Newsletter. 20 (2): 15–19. doi:10.1145/360262.360268. S2CID 9417766.
- ↑ Cock, Peter JA; Antao, Tiago; Chang, Jeffery T; Chapman, Brad A; Cox, Cymon J; Dalke, Andrew; Friedberg, Iddo; Hamelryck, Thomas; Kauff, Frank; Wilczynski, Bartek; de Hoon, Michiel JL (2009 թ․ մարտի 20). «Biopython: freely available Python tools for computational molecular biology and bioinformatics». Bioinformatics. 25 (11): 1422–3. doi:10.1093/bioinformatics/btp163. PMC 2682512. PMID 19304878.
- ↑ 3,0 3,1 Refer to the Biopython website for other papers describing Biopython, and a list of over one hundred publications using/citing Biopython.
- ↑ Mangalam, Harry (September 2002). «The Bio* toolkits—a brief overview». Briefings in Bioinformatics. 3 (3): 296–302. doi:10.1093/bib/3.3.296. PMID 12230038.
- ↑ 5,0 5,1 Chapman, Brad (11 March 2004), The Biopython Project: Philosophy, functionality and facts (PDF), Վերցված է 11 September 2014-ին
- ↑ List of Biopython contributors, Արխիվացված է օրիգինալից 11 September 2014-ին, Վերցված է 11 September 2014-ին
- ↑ Knight, R; Maxwell, P; Birmingham, A; Carnes, J; Caporaso, J. G.; Easton, B. C.; Eaton, M; Hamady, M; Lindsay, H; Liu, Z; Lozupone, C; McDonald, D; Robeson, M; Sammut, R; Smit, S; Wakefield, M. J.; Widmann, J; Wikman, S; Wilson, S; Ying, H; Huttley, G. A. (2007). «Py Cogent: A toolkit for making sense from sequence». Genome Biology. 8 (8): R171. doi:10.1186/gb-2007-8-8-r171. PMC 2375001. PMID 17708774.
{cite journal}: CS1 սպաս․ չպիտակված ազատ DOI (link) - ↑ Daley, Chris, Biopython 1.77 released, Վերցված է 6 October 2021-ին
- ↑ Chang, Jeff; Chapman, Brad; Friedberg, Iddo; Hamelryck, Thomas; de Hoon, Michiel; Cock, Peter; Antao, Tiago; Talevich, Eric; Wilczynski, Bartek (29 May 2014), Biopython Tutorial and Cookbook, Վերցված է 28 August 2014-ին
- ↑ Zmasek, Christian M; Zhang, Qing; Ye, Yuzhen; Godzik, Adam (2007 թ․ հոկտեմբերի 24). «Surprising complexity of the ancestral apoptosis network». Genome Biology. 8 (10): R226. doi:10.1186/gb-2007-8-10-r226. PMC 2246300. PMID 17958905. Արխիվացված է օրիգինալից 2014 թ․ հոկտեմբերի 10-ին. Վերցված է 2014 թ․ սեպտեմբերի 9-ին.
{cite journal}: CS1 սպաս․ չպիտակված ազատ DOI (link) - ↑ Talevich, Eric; Invergo, Brandon M; Cock, Peter JA; Chapman, Brad A (2012 թ․ օգոստոսի 21). «Bio.Phylo: A unified toolkit for processing, analyzing and visualizing phylogenetic trees in Biopython». BMC Bioinformatics. 13 (209): 209. doi:10.1186/1471-2105-13-209. PMC 3468381. PMID 22909249.
{cite journal}: CS1 սպաս․ չպիտակված ազատ DOI (link) - ↑ «Klebsiella pneumoniae strain KPS77 plasmid pKPS77, complete sequence». NCBI. Վերցված է 2014 թ․ սեպտեմբերի 10-ին.
- ↑ Pritchard, Leighton; White, Jennifer A; Birch, Paul RJ; Toth, Ian K (March 2006). «GenomeDiagram: a python package for the visualization of large-scale genomic data». Bioinformatics. 22 (5): 616–617. doi:10.1093/bioinformatics/btk021. PMID 16377612.
- ↑ Rousset, François (January 2008). «GENEPOP'007: a complete re-implementation of the GENEPOP software for Windows and Linux». Molecular Ecology Resources. 8 (1): 103–106. doi:10.1111/j.1471-8286.2007.01931.x. PMID 21585727. S2CID 25776992.
- ↑ Excoffier, Laurent; Foll, Matthieu (2011 թ․ մարտի 1). «fastsimcoal: a continuous-time coalescent simulator of genomic diversity under arbitrarily complex evolutionary scenarios». Bioinformatics. 27 (9): 1332–1334. doi:10.1093/bioinformatics/btr124. PMID 21398675.