Memory T cell
Memory T cells are a subset of T lymphocytes that might have some of the same functions as memory B cells. Their lineage is unclear.
Function
Antigen-specific memory T cells specific to viruses or other microbial molecules can be found in both central memory T cells (TCM) and effector memory T cells (TEM) subsets. Although most information is currently based on observations in the cytotoxic T cells (CD8-positive) subset, similar populations appear to exist for both the helper T cells (CD4-positive) and the cytotoxic T cells. Primary function of memory cells is augmented immune response after reactivation of those cells by reintroduction of relevant pathogen into the body. It is important to note that this field is intensively studied and some information may not be available as of yet.
- Central memory T cells (TCM): TCM lymphocytes have several attributes in common with stem cells, the most important being the ability of self-renewal, mainly because of high level of phosphorylation on key transcription factor STAT5. In mice, TCM proved to confer more powerful immunity against viruses,[1] bacteria[1] and cancer cells,[2] compared to TEM lymphocytes in several experimental models.
- Effector memory T cells (TEM): TEM and TEMRA lymphocytes are primarily active as the CD8 variants, thus being mainly responsible for cytotoxic action against pathogens.[3]
- Tissue-resident memory T cell (TRM): Because TRM lymphocytes are present over long periods of time in tissues, or more importantly, barrier tissues (epithelium for example), they are crucial for quick response to barrier breach and response to any relevant pathogen present. One mechanism used by TRM to restrict pathogens is the secretion of granzyme B.[4][5]
- Stem cell-like memory T cells (TSCM): Those lymphocytes are capable of self-renewal as are the TCM lymphocytes and are also capable of generating both the TCM and TEM subpopulations.[6] Presence of this population in humans is currently under investigation.
- Virtual memory T cell (TVM): As of now, the only function apparent in TVM cells is production of various cytokines,[7][8] but there are speculations about their influence in subduing unwanted immunological states and their usage in treating autoimmune disorders.[9]
Homeostatic maintenance
Clones of memory T cells expressing a specific T cell receptor can persist for decades in our body. Since memory T cells have shorter half-lives than naïve T cells do, continuous replication and replacement of old cells are likely involved in the maintenance process.[3] Currently, the mechanism behind memory T cell maintenance is not fully understood. Activation through the T cell receptor may play a role.[3] It is found that memory T cells can sometimes react to novel antigens, potentially caused by intrinsic the diversity and breadth of the T cell receptor binding targets.[3] These T cells could cross-react to environmental or resident antigens in our bodies (like bacteria in our gut) and proliferate. These events would help maintain the memory T cell population.[3] The cross-reactivity mechanism may be important for memory T cells in the mucosal tissues since these sites have higher antigen density.[3] For those resident in blood, bone marrow, lymphoid tissues, and spleen, homeostatic cytokines (including IL-17 and IL-15) or major histocompatibility complex II (MHCII) signaling may be more important.[3]
Lifetime overview
Memory T cells undergo different changes and play different roles in different life stages for humans. At birth and early childhood, T cells in the peripheral blood are mainly naïve T cells.[10] Through frequent antigen exposure, the population of memory T cells accumulates. This is the memory generation stage, which lasts from birth to about 20–25 years old when our immune system encounters the greatest number of new antigens.[3][10] During the memory homeostasis stage that comes next, the number of memory T cells plateaus and is stabilized by homeostatic maintenance.[10] At this stage, the immune response shifts more towards maintaining homeostasis since few new antigens are encountered.[10] Tumor surveillance also becomes important at this stage.[10] At later stages of life, at about 65–70 years of age, immunosenescence stage comes, in which stage immune dysregulation, decline in T cell function and increased susceptibility to pathogens are observed.[3][10]
Lineage debate
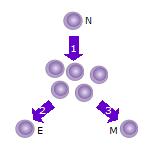
- After the naive T cell (N) encounters an antigen it becomes activated and begins to proliferate (divide) into many clones or daughter cells.
- Some of the T cell clones will differentiate into effector T cells (E) that will perform the function of that cell (e.g. produce cytokines in the case of helper T cells or invoke cell killing in the case of cytotoxic T cells).
- Some of the cells will form memory T cells (M) that will survive in an inactive state in the host for a long period of time until they re-encounter the same antigen and reactivate.
As of April 2020, the lineage relationship between effector and memory T cells is unclear.[11][12][13] Two competing models exist. One is called the On-Off-On model.[12] When naive T cells are activated by T cell receptor (TCR) binding to antigen and its downstream signaling pathway, they actively proliferate and form a large clone of effector cells. Effector cells undergo active cytokine secretion and other effector activities.[11] After antigen clearance, some of these effector cells form memory T cells, either in a randomly determined manner or are selected based on their superior specificity.[11] These cells would reverse from the active effector role to a state more similar to naive T cells and would be "turned on" again upon the next antigen exposure.[13] This model predicts that effector T cells can transit into memory T cells and survive, retaining the ability to proliferate.[11] It also predicts that certain gene expression profiles would follow the on-off-on pattern during naive, effector, and memory stages.[13] Evidence supporting this model includes the finding of genes related to survival and homing that follow the on-off-on expression pattern, including interleukin-7 receptor alpha (IL-7Rα), Bcl-2, CD26L, and others.[13]

In this model, memory T cells generate effector T cells, not the other way around.
The other model is the developmental differentiation model.[12] This model argues that effector cells produced by the highly activated naive T cells would all undergo apoptosis after antigen clearance.[11] Memory T cells are instead produced by naive T cells that are activated but never entered with full strength into the effector stage.[11] The progeny of memory T cells are not fully activated because they are not as specific to the antigen as the expanding effector T cells. Studies looking at cell division history found that the length of telomere and activity of telomerase were reduced in effector T cells compared to memory T cells, which suggests that memory T cells did not undergo as much cell division as effector T cells, which is inconsistent with the On-Off-On model.[11] Repeated or chronic antigenic stimulation of T cells, like HIV infection, would induce elevated effector functions but reduce memory.[12] It was also found that massively proliferated T cells are more likely to generate short-lived effector cells, while minimally proliferated T cells would form more long-lived cells.[11]
Epigenetic modifications
Epigenetic modifications are involved in the change from naive T-cells.[14] For example, in CD4+ memory T cells, positive histone modifications mark key cytokine genes that are up-regulated during the secondary immune response, including IFNγ, IL4, and IL17A.[14] Some of these modifications persisted after antigen clearance, establishing an epigenetic memory that allows a faster activation upon re-encounter with the antigen.[14] For CD8+ memory T cells, certain effector genes, such as IFNγ, would not be expressed but they are transcriptionally poised for fast expression upon activation.[14] Additionally, the enhancement of expression for certain genes also depends on the strength of the initial TCR signaling for the progeny of memory T cells, which is correlated to the regulatory element activation that directly changes gene expression level.[14]
Sub-populations
Historically, memory T cells were thought to belong to either the effector (TEM cells) or central memory (TCM cells) subtypes, each with its own distinguishing set of cell surface markers (see below).[15] Subsequently, numerous additional populations of memory T cells were discovered including tissue-resident memory T (TRM) cells, stem memory TSCM cells, and virtual memory T cells. The single unifying theme for all memory T cell subtypes is that they are long-lived and can quickly expand to large numbers of effector T cells upon re-exposure to their cognate antigen. By this mechanism, they provide the immune system with "memory" against previously encountered pathogens. Memory T cells may be either CD4+ or CD8+ and usually express CD45RO and at the same time lack CD45RA.[16]
Memory T cell subtypes
- Central memory T cells (TCM cells) express CD45RO, C-C chemokine receptor type 7 (CCR7), and L-selectin (CD62L). Central memory T cells also have intermediate to high expression of CD44. This memory subpopulation is commonly found in the lymph nodes and in the peripheral circulation.
- Effector memory T cells (TEM cells) express CD45RO but lack expression of CCR7 and L-selectin. They also have intermediate to high expression of CD44. Because these memory T cells lack the CCR7 lymph node-homing receptors they are found in the peripheral circulation and tissues.[17] TEMRA stands for terminally differentiated effector memory cells re-expressing CD45RA, which is a marker usually found on naive T cells.[18]
- Peripheral memory T cells (TPM cells) subtype was identified based on intermediate CX3CR1 expression. These cells can migrate to the tissues from blood and traffic to the lymph nodes in a CD62L-independent manner, in order to survey the tissues. [19]
- Tissue-resident memory T cells (TRM) occupy tissues (skin, lung, gastrointestinal tract, etc.) without recirculating. Some cell surface markers that have been associated with TRM are CD69 and integrin αeβ7 (CD103).[20] However, it is worth noticing that TRM cells found in different tissues express different sets of cell surface markers.[20] While CD103+ TRM cells are found to be restrictedly localized to epithelial and neuronal tissues, TRM cells localized in salivary glands, pancreas, and female reproductive tracts in mice express neither CD69 nor CD103.[20][21] TRM cells are thought to play a major role in protective immunity against pathogens.[5][22] Studies have also suggested a dual role for TRM cells in protection and regulation.[10] Compared to TEM cells, TRM cells secrete higher levels of protective-immunity-related cytokines and express lower levels of the proliferation marker Ki67.[10] It was proposed that these characteristics may help with the long-term maintenance of TRM cells, as well as keeping a balance between quick response to antigen invasion and avoidance of unnecessary tissue damage.[10] Dysfunctional TRM cells have been implicated in autoimmune diseases, such as psoriasis, rheumatoid arthritis, and inflammatory bowel disease.[22] Specific to TRM lymphocytes are genes involved in lipid metabolism, being highly active, roughly 20- to 30-fold more active than in other types of T-cells.[22]
- Virtual memory T cells (TVM) differ from the other memory subsets in that they do not originate following a strong clonal expansion event. Thus, although this population as a whole is abundant within the peripheral circulation, individual virtual memory T cell clones reside at relatively low frequencies. One theory is that homeostatic proliferation gives rise to this T cell population. Although CD8 virtual memory T cells were the first to be described,[23] it is now known that CD4 virtual memory cells also exist.[24]
There have been numerous other subpopulations of memory T cells suggested. Investigators have studied Stem memory TSCM cells. Like naive T cells, TSCM cells are CD45RO−, CCR7+, CD45RA+, CD62L+ (L-selectin), CD27+, CD28+, and IL-7Rα+, but they also express large amounts of CD95, IL-2Rβ, CXCR3, and LFA-1, and show numerous functional attributes distinctive of memory cells.[6]
TCR-independent (bystander) activation
T cells possess the ability to be activated independently of their cognate antigen stimulation, i.e. without TCR stimulation. At early stages of infection, T cells specific for unrelated antigen are activated only by the presence of inflammation. This happens in the inflammatory milieu resulting from microbial infection, cancer or autoimmunity in both mice and humans and occurs locally as well as systematically [25][26][27][28][29] . Moreover, bystander activated T cells can migrate to the site of infection, due to increased CCR5 expression.[26]
This phenomenon was observed predominantly in memory CD8+ T cells, which have lower sensitivity to cytokine stimulation, compared to their naive counterparts and get activated in this manner more easily.[25] Virtual memory CD8+ T cells also display heightened sensitivity to cytokine-induced activation in mouse models, but this was not directly demonstrated in humans.[26] Conversely, TCR-independent activation of naive CD8+ T cells remains controversial.[26][28]
Apart from infections, bystander activation also plays an important role in antitumor immunity.[30] In human cancerous tissues, a high number of virus-specific, not tumor-specific, CD8+ T cells was detected.[30] This type of activation is considered to be beneficial for the host in terms of cancer clearance efficiency.[26]
Drivers of bystander activation
The major drivers of bystander activation are cytokines, such as IL-15, IL-18, IL-12 or type I IFNs, often working synergistically.[25][26][28][29] IL-15 is responsible for cytotoxic activity of bystander-activated T cells. It induces the NKG2D (a receptor typically expressed on NK cells) expression on memory CD8+ T cells, leading to innate-like cytotoxicity, i.e. recognition of NKG2D ligands as indicators of infection, cell stress and cell transformation as well as destruction of altered cells in an NK-like manner.[25][26][28][29] TCR activation was shown to abrogate IL-15 mediated NKG2D expression on T cells.[28][29] Additionally, IL-15 induces expression of cytolytic molecules, cell expansion and enhances the cell response to IL-18.[25][26][29] IL-18 is another cytokine involved in this process, typically acting in synergy with IL-12, enhancing the differentiation of memory T cells into effector cells, i.e. it induces IFN-γ production and cell proliferation.[25][26][29] Toll-like receptors (TLRs), especially TLR2, have been linked to TCR-independent activation of CD8+ T cells upon bacterial infection as well.[25][29]
Bystander activation of CD4+ T cells
Despite TCR-independent activation being studied more extensively in CD8+ T cells, there’s a clear evidence of this phenomenon occurring in CD4+ T cells. However, it’s considered to be less efficient, presumably due to lower CD122 (also known as IL2RB or IL15RB) expression.[31][32] Similarly to their CD8+ counterparts, memory and effector CD4+ T cells exhibit increased sensitivity to TCR-independent activation.[26][32] IL-1β, synergistically with IL-12 and IL-23, stimulates memory CD4+ T cells and drives Th17 response.[32] Moreover, IL-18, IL-12 and IL-27 induce cytokine expression in effector and memory CD4+ T cells[32] and IL-2 is considered to be a strong activation inducer of CD4+ T cells that can replace TCR stimulation even in naive cells.[32] TLR2 was also reported to be present on memory CD4+ T cells, which respond to their agonist by IFNγ production, even without TCR stimulation.[32]
Role in pathogenicity
Bystander activation plays role in the elimination of the spread of infection in its early stages and helps in tumor clearance. However, this type of activation can also have deleterious outcome, especially in chronic infections and autoimmune diseases.[26][27][28][29] Liver injury during chronic Hepatitis B virus infection is a result of non-HBV-specific CD8+ T cell infiltration into the tissue.[26] A similar situation occurs during the acute Hepatitis A virus infection[26] and activated virus unrelated CD4+ T cells contribute to ocular lesions in Herpes Simplex Virus infections.[26][32]
Increased IL-15 expression and subsequent excessive NKG2D expression was linked to exacerbation of some autoimmune disorders, such as, type I diabetes, multiple sclerosis and inflammatory bowel diseases, for instance Crohn’s disease and celiac disease.[25] Furthermore, enhanced TLR2 expression was observed in joints, cartilage and bones of rheumatoid arthritis patients and the presence of its ligand, peptidoglycan, was detected in their synovial fluid.[25]
References
- ^ a b Wherry EJ, Teichgräber V, Becker TC, Masopust D, Kaech SM, Antia R, et al. (March 2003). "Lineage relationship and protective immunity of memory CD8 T cell subsets". Nature Immunology. 4 (3): 225–34. doi:10.1038/ni889. PMID 12563257. S2CID 7209417.
- ^ Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. (July 2005). "Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells". Proceedings of the National Academy of Sciences of the United States of America. 102 (27): 9571–6. Bibcode:2005PNAS..102.9571K. doi:10.1073/pnas.0503726102. PMC 1172264. PMID 15980149.
- ^ a b c d e f g h i Farber DL, Yudanin NA, Restifo NP (January 2014). "Human memory T cells: generation, compartmentalization and homeostasis". Nature Reviews. Immunology. 14 (1): 24–35. doi:10.1038/nri3567. PMC 4032067. PMID 24336101.
- ^ Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR (May 2009). "Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus". Nature Immunology. 10 (5): 524–30. doi:10.1038/ni.1718. PMID 19305395. S2CID 24388.
- ^ a b Shin H, Iwasaki A (September 2013). "Tissue-resident memory T cells". Immunological Reviews. 255 (1): 165–81. doi:10.1111/imr.12087. PMC 3748618. PMID 23947354.
- ^ a b Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. (September 2011). "A human memory T cell subset with stem cell-like properties". Nature Medicine. 17 (10): 1290–7. doi:10.1038/nm.2446. PMC 3192229. PMID 21926977.
- ^ White JT, Cross EW, Kedl RM (June 2017). "+ T cells: where they come from and why we need them". Nature Reviews. Immunology. 17 (6): 391–400. doi:10.1038/nri.2017.34. PMC 5569888. PMID 28480897.
- ^ Lee JY, Hamilton SE, Akue AD, Hogquist KA, Jameson SC (August 2013). "Virtual memory CD8 T cells display unique functional properties". Proceedings of the National Academy of Sciences of the United States of America. 110 (33): 13498–503. Bibcode:2013PNAS..11013498L. doi:10.1073/pnas.1307572110. PMC 3746847. PMID 23898211.
- ^ Drobek A, Moudra A, Mueller D, Huranova M, Horkova V, Pribikova M, et al. (July 2018). "Strong homeostatic TCR signals induce formation of self-tolerant virtual memory CD8 T cells". The EMBO Journal. 37 (14). doi:10.15252/embj.201798518. PMC 6043851. PMID 29752423.
- ^ a b c d e f g h i Kumar BV, Connors TJ, Farber DL (February 2018). "Human T Cell Development, Localization, and Function throughout Life". Immunity. 48 (2): 202–213. doi:10.1016/j.immuni.2018.01.007. PMC 5826622. PMID 29466753.
- ^ a b c d e f g h Restifo NP, Gattinoni L (October 2013). "Lineage relationship of effector and memory T cells". Current Opinion in Immunology. Special section: Systems biology and bioinformatics / Immunogenetics and transplantation. 25 (5): 556–63. doi:10.1016/j.coi.2013.09.003. PMC 3858177. PMID 24148236.
- ^ a b c d Henning AN, Roychoudhuri R, Restifo NP (May 2018). "+ T cell differentiation". Nature Reviews. Immunology. 18 (5): 340–356. doi:10.1038/nri.2017.146. PMC 6327307. PMID 29379213.
- ^ a b c d Youngblood B, Hale JS, Ahmed R (July 2013). "T-cell memory differentiation: insights from transcriptional signatures and epigenetics". Immunology. 139 (3): 277–84. doi:10.1111/imm.12074. PMC 3701173. PMID 23347146.
- ^ a b c d e Schmidl C, Delacher M, Huehn J, Feuerer M (September 2018). "Epigenetic mechanisms regulating T-cell responses". The Journal of Allergy and Clinical Immunology. 142 (3): 728–743. doi:10.1016/j.jaci.2018.07.014. PMID 30195378.
- ^ Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A (October 1999). "Two subsets of memory T lymphocytes with distinct homing potentials and effector functions". Nature. 401 (6754): 708–12. Bibcode:1999Natur.401..708S. doi:10.1038/44385. PMID 10537110. S2CID 4378970.
- ^ Akbar AN, Terry L, Timms A, Beverley PC, Janossy G (April 1988). "Loss of CD45R and gain of UCHL1 reactivity is a feature of primed T cells". Journal of Immunology. 140 (7): 2171–8. doi:10.4049/jimmunol.140.7.2171. PMID 2965180. S2CID 22340282.
- ^ Willinger T, Freeman T, Hasegawa H, McMichael AJ, Callan MF (November 2005). "Molecular signatures distinguish human central memory from effector memory CD8 T cell subsets". Journal of Immunology. 175 (9): 5895–903. doi:10.4049/jimmunol.175.9.5895. hdl:20.500.11820/f28e936e-a6a7-4f06-bdc9-79a1355c5f02. PMID 16237082.
- ^ Koch S, Larbi A, Derhovanessian E, Ozcelik D, Naumova E, Pawelec G (July 2008). "Multiparameter flow cytometric analysis of CD4 and CD8 T cell subsets in young and old people". Immunity & Ageing. 5 (6): 6. doi:10.1186/1742-4933-5-6. PMC 2515281. PMID 18657274.
- ^ Gerlach, Carmen; Moseman, E. Ashley; Loughhead, Scott M.; Alvarez, David; Zwijnenburg, Anthonie J.; Waanders, Lisette; Garg, Rohit; de la Torre, Juan C.; von Andrian, Ulrich H. (December 2016). "The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets with Distinct Roles in Immune Surveillance and Homeostasis". Immunity. 45 (6): 1270–1284. doi:10.1016/j.immuni.2016.10.018. PMC 5177508. PMID 27939671.
- ^ a b c Mueller SN, Mackay LK (February 2016). "Tissue-resident memory T cells: local specialists in immune defence". Nature Reviews. Immunology. 16 (2): 79–89. doi:10.1038/nri.2015.3. PMID 26688350. S2CID 3155731.
- ^ Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyártó BZ, et al. (May 2015). "Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance". Cell. 161 (4): 737–49. doi:10.1016/j.cell.2015.03.031. PMC 4426972. PMID 25957682.
- ^ a b c "Study highlights possible Achilles' heel in key immune memory cells".
- ^ Lee YJ, Jameson SC, Hogquist KA (February 2011). "Alternative memory in the CD8 T cell lineage". Trends in Immunology. 32 (2): 50–6. doi:10.1016/j.it.2010.12.004. PMC 3039080. PMID 21288770.
- ^ Marusina AI, Ono Y, Merleev AA, Shimoda M, Ogawa H, Wang EA, et al. (February 2017). "+ virtual memory: Antigen-inexperienced T cells reside in the naïve, regulatory, and memory T cell compartments at similar frequencies, implications for autoimmunity". Journal of Autoimmunity. 77: 76–88. doi:10.1016/j.jaut.2016.11.001. PMC 6066671. PMID 27894837.
- ^ a b c d e f g h i Whiteside, Sarah K.; Snook, Jeremy P.; Williams, Matthew A.; Weis, Janis J. (December 2018). "Bystander T Cells: A Balancing Act of Friends and Foes". Trends in Immunology. 39 (12): 1021–1035. doi:10.1016/j.it.2018.10.003. PMC 6269193. PMID 30413351.
- ^ a b c d e f g h i j k l m n Lee, Hoyoung; Jeong, Seongju; Shin, Eui-Cheol (January 2022). "Significance of bystander T cell activation in microbial infection". Nature Immunology. 23 (1): 13–22. doi:10.1038/s41590-021-00985-3. ISSN 1529-2908. PMID 34354279. S2CID 236933989.
- ^ a b Pacheco, Yovana; Acosta-Ampudia, Yeny; Monsalve, Diana M.; Chang, Christopher; Gershwin, M. Eric; Anaya, Juan-Manuel (September 2019). "Bystander activation and autoimmunity". Journal of Autoimmunity. 103: 102301. doi:10.1016/j.jaut.2019.06.012. PMID 31326230. S2CID 198133084.
- ^ a b c d e f Maurice, Nicholas J.; Taber, Alexis K.; Prlic, Martin (2021-02-01). "The Ugly Duckling Turned to Swan: A Change in Perception of Bystander-Activated Memory CD8 T Cells". The Journal of Immunology. 206 (3): 455–462. doi:10.4049/jimmunol.2000937. ISSN 0022-1767. PMC 7839146. PMID 33468558.
- ^ a b c d e f g h Kim, Tae-Shin; Shin, Eui-Cheol (December 2019). "The activation of bystander CD8+ T cells and their roles in viral infection". Experimental & Molecular Medicine. 51 (12): 1–9. doi:10.1038/s12276-019-0316-1. ISSN 1226-3613. PMC 6906361. PMID 31827070.
- ^ a b Borras, DM, Verbandt, S, Ausserhofer, M, et al. (November 2023). "Single cell dynamics of tumor specificity vs bystander activity in CD8+ T cells define the diverse immune landscapes in colorectal cancer". Cell. Discovery. 9 (114): 114. doi:10.1038/s41421-023-00605-4. PMC 10652011. PMID 37968259.
- ^ Boyman, Onur (April 2010). "Bystander activation of CD4 + T cells: HIGHLIGHTS". European Journal of Immunology. 40 (4): 936–939. doi:10.1002/eji.201040466. PMID 20309907. S2CID 7918378.
- ^ a b c d e f g Lee, Hong-Gyun; Cho, Min-Zi; Choi, Je-Min (August 2020). "Bystander CD4+ T cells: crossroads between innate and adaptive immunity". Experimental & Molecular Medicine. 52 (8): 1255–1263. doi:10.1038/s12276-020-00486-7. ISSN 1226-3613. PMC 8080565. PMID 32859954.