Talasemia
| thalassaemia | |
 Rozmaz krwi u chorego z delta-beta talasemią | |
| Klasyfikacje | |
| ICD-10 | |
|---|---|
Talasemia (niedokrwistość tarczowatokrwinkowa, łac. thalassaemia, ang. thalassemia) – ilościowe zaburzenia syntezy hemoglobiny, spowodowane wrodzonym defektem biosyntezy łańcuchów globiny. Najczęściej zaburzenia dotyczą ekspresji alfa-globiny (alfa-talasemia) lub beta-globiny (beta-talasemia), choć istnieją także talasemie związane z obniżoną syntezą innych globin np. delta-globiny, gamma-globiny. Zaburzenia w biosyntezie globin spowodowane są mutacjami w kodujących je genach lub ich elementach regulatorowych.
Talasemia określana jest różnymi nazwami: „niedokrwistość śródziemnomorska" (thalassa — "morze" + haima — "krew"; z greki: "z okolic morza" — nazwa związana z występowaniem choroby w rejonie Morza Śródziemnego) lub „niedokrwistość tarczowatokrwinkowa" (nazwa związana z wyglądem krwinek), a ciężka (major) postać beta- talasemii to inaczej „niedokrwistość Cooleya", od nazwiska pediatry, który ją opisał w 1925 roku.
"Ciężkość" talasemii związana jest najczęściej z: rodzajem mutacji sprawczej, rodzajem dziedziczenia mutacji (np. postać heterozygotyczna, homozygotyczna, czy też złożona heterozygota), współdziedziczeniem innych talasemii, wariantów hemoglobin oraz innymi czynnikami.
Alfa-talasemia dzieli się na cztery główne rodzaje: tzw. cichy nosiciel (najłagodniejsza postać), cecha alfa-talasemii (tzw. trait), choroba HbH oraz HbBart’s hydrops fetalis (najcięższa postać alfa-talasemii).
Beta-talasemia dzieli się na trzy główne rodzaje: minor (tzw. cecha beta-talasemii), intermedia, major (najcięższa postać beta-talasemii).
Objawami talasemii są m.in. ubytki kości[1], anemia, powiększenia śledziony, wątroby, węzłów chłonnych, pojawianie się kamieni nerkowych i bóle w kończynach dolnych[2].
Epidemiologia
Dwie główne postaci talasemii tzn. alfa- i beta-talasemia są szczególnie szeroko rozpowszechnione w niektórych regionach świata. Dotyczy to głównie obszarów basenu Morza Śródziemnego, Bliskiego Wschodu, Indii i Azji Południowo-Wschodniej, a także niektórych regionów Afryki. Obecnie uważa się, że obecność talasemii w populacjach ludności tych rejonów świata stanowiła selektywną ochronę przed malarią, gdyż taki wpływ potwierdziły badania[3][4]. Migracje ludności spowodowały jednak, że talasemie są obecnie również w rejonach świata gdzie pierwotnie nie występowały np. obu Amerykach, Australii i różnych rejonach Europy.
Symptomy
Chociaż są badacze twierdzący, że talasemia beta minor jest asymptomatyczna[5], to większość doniesień mówi o objawach, do których należy męczliwość i mięśnioból[6].
Dziedziczenie
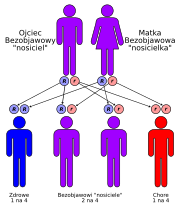
Zarówno alfa-, jak i beta-talasemia w większości przypadków są dziedziczone w sposób autosomalny recesywny, choć w przypadku beta-talasemii zdarzają się też dominujące typy mutacji.
Diagnostyka
Ogólna diagnostyka talasemii oparta jest na m.in.:
- Wywiadzie lekarskim, pozwalającym m.in. na wykrycie ewentualnych rodzinnych (genetycznych) uwarunkowań choroby. Istotne jest poznanie grupy etnicznej pacjenta oraz obserwacja objawów klinicznych.
- Badaniu morfologii krwi w celu wykrycia niedokrwistości (najczęściej spadek stężenia hemoglobiny). Ponadto najczęściej stwierdza się we krwi mikrocytozę z obniżeniem MCH. Wykonanie rozmazu krwi jest także istotne, szczególnie w cięższych postaciach talasemii. Niektóre łagodne postaci talasemii mogą jednak wykazywać parametry krwi (morfologia) w granicach normy. W przypadku cięższych postaci talasemii bardzo pomocne jest także określenie czasu wystąpienia pierwszych objawów oraz zależność od transfuzji.
- Badaniu jakościowym i ilościowym hemoglobin (także wariantów hemoglobin). W przypadku talasemii głównie chodzi tutaj o sprawdzenie obecności i oznaczenie poziomu HbA2, HbF i HbA np.w heterozygotycznej postaci beta-talasemii występuje zmniejszenie wytwarzania łańcucha β-globiny, co prowadzi do spadku HbA i zwiększenia ilości HbA2; ważne jest też badanie HbF - u chorych z tą postacią talasemii może występować kompensacyjny wzrost produkcji γ-globiny, co powoduje zwiększoną zawartość HbF. Badania ilościowe hemoglobin (także wariantów hemoglobin) oparte są obecnie na diagnostyce przy użyciu elektroforezy kapilarnej (CE), metod chromatograficznych np. HPLC, chromatografii mikrokolumienkowej oraz innych metod diagnostycznych. Metody jakościowe np. elektroforeza hemoglobin w środowisku zasadowym i kwaśnym (nie należy mylić z elektroforezą kapilarną) umożliwiają m.in. wykrycie wariantów hemoglobin, które mogą współtowarzyszyć talasemiom i dodatkowo zaburzać obraz diagnostyczny i kliniczny pacjenta.
- Niezbędna jest także diagnostyka molekularna, która pozwoli na jednoznaczne określenie mutacji sprawczej np. rozróżnienie beta-zero czy też beta-plus talasemii.
- W przypadku alfa-talasemii badania genetyczne są niezbędne do diagnostyki tej niedokrwistości. Pomocniczo, obniżony lub pozostający na dolnej granicy normy poziom HbA2 może wskazywać na cechę alfa-talasemii, choć w dużej części przypadków alfa-talasemii wartości HbA2 mogą nie odbiegać od normy, a dalsza diagnostyka musi się opierać na metodach biologii molekularnej. Jedynie w przypadku choroby HbH wartość HbA2 może być znacznie obniżona. Szczególnie w przypadku cięższych postaci alfa-talasemii pomocne są także badania (np. HPLC, elektroforeza kapilarna, elektroforeza hemoglobin) pozwalające na detekcję m.in. HbH i/lub HbBart's.
Klasyfikacja ICD10
| kod ICD10 | nazwa choroby |
|---|---|
| ICD-10: D56 | Talasemia |
| ICD-10: D56.0 | Talasemia alfa |
| ICD-10: D56.1 | Talasemia beta |
| ICD-10: D56.2 | Talasemia delta-beta |
| ICD-10: D56.3 | Cecha talasemii |
| ICD-10: D56.4 | Dziedziczna przetrwała hemoglobina płodowa (HPPH) |
| ICD-10: D56.8 | Inne talasemie |
| ICD-10: D56.9 | Nieokreślona talasemia |
Przypisy
- ↑ Maria G Vogiatzi i inni, Bone Disease in Thalassemia: A Frequent and Still Unresolved Problem, „Journal of Bone and Mineral Research”, 24 (3), 2017, s. 543–557, DOI: 10.1359/jbmr.080505, ISSN 0884-0431, PMID: 18505376, PMCID: PMC3276604 [dostęp 2017-02-02].
- ↑ Renzo Galanello, Raffaella Origa, Beta-thalassemia, „Orphanet Journal of Rare Diseases”, 5, 2010, s. 11, DOI: 10.1186/1750-1172-5-11, ISSN 1750-1172, PMID: 20492708, PMCID: PMC2893117 [dostęp 2017-02-02].
- ↑ Sammy Wambua i inni, The Effect of α +-Thalassaemia on the Incidence of Malaria and Other Diseases in Children Living on the Coast of Kenya, „PLoS Medicine”, 3 (5), 2017, DOI: 10.1371/journal.pmed.0030158, ISSN 1549-1277, PMID: 16605300, PMCID: PMC1435778 [dostęp 2017-02-02].
- ↑ G. Modiano i inni, Protection against malaria morbidity: Near-fixation of the α-thalassemia gene in a Nepalese population, „American Journal of Human Genetics”, 48 (2), 1991, s. 390–397, ISSN 0002-9297, PMID: 1990845, PMCID: PMC1683029 [dostęp 2017-02-02].
- ↑ Renzo Galanello, Raffaella Origa, Beta-thalassemia, „Orphanet Journal of Rare Diseases”, 5, 2010, s. 11, DOI: 10.1186/1750-1172-5-11, ISSN 1750-1172, PMID: 20492708, PMCID: PMC2893117 [dostęp 2019-07-21].
- ↑ Seyed Mohammad Bagher Tabei i inni, Determining and surveying the role of carnitine and folic acid to decrease fatigue in β-thalassemia minor subjects, „Pediatric Hematology and Oncology”, 30 (8), 2013, s. 742–747, DOI: 10.3109/08880018.2013.771388, ISSN 1521-0669, PMID: 23458634 [dostęp 2019-07-21].
Linki zewnętrzne
Bibliografia
- Paweł Turowski, Małgorzata Uhrynowska, Ewa Brojer. Talasemie – patofizjologia, podstawy molekularne i diagnostyka. „Hematologia”, 2013.
- Andrzej Szczeklik (red.): Choroby wewnętrzne. T. II. Wydawnictwo Medycyna Praktyczna, 2005, s. 1455-1456. ISBN 83-7430-069-8.